¶ Systemoppsett for relativ FEP – fra PDB/SMILES til CHARMM-GUI-prosjekt (for senere bruk i FEP)
Forfatter: Majd Awad
Tilknyttet gruppe: LIPCHEM – Legemiddelkjemi
Publisert: September 2025
Lisens: CC BY-SA 4.0
Omfang: Denne veiledningen dekker kun systembygging (protein, ligand(er), membran, ioner, solvatisering) i CHARMM-GUI for senere bruk i relativ FEP. Selve kjøring av FEP i NAMD og analyse dekkes i egne guider.
¶ 🎯 Mål
- Forberede et komplett, simulering-klart protein–ligand-system for relativ FEP.
- Standardisere input (PDB, SDF/MOL2), membranoppsett og ionisering slik at neste steg (FEP) blir reproduserbart.
¶ Forberedelser
| Ressurs | Format | Hvor/hvordan |
|---|---|---|
| Protein | .pdb |
RCSB PDB (eks. FPR2, PDB: 7T6S) |
| Referanseligand (med kjent affinitet) | .sdf eller .mol2 |
Docking/eksperiment; evt. SDF fra RCSB hvis posisjon kjent |
| Målligander (analoger) | .sdf |
Fra SMILES → SDF (ChemDraw/Open Babel/Maestro) |
| Byggeverktøy | CHARMM-GUI | CHARMM-GUI Free Energy Calculator – Relative Ligand Binder |
¶ 1) Generer ligandfiler (SDF/MOL2)
¶ Metode A – ChemDraw (kan feile for komplekse strukturer)
- SMILES →
Edit → Paste Special → SMILES Structure → Clean Structure(rydde opp)- Sjekk protonering/ladning (pH-relevant)
File → Save As…→ SDF eller MOL2
🖼️ Bilde: ChemDraw “Clean Structure”.
¶ Metode B – Open Babel (anbefalt)
Installer Open Babel og kjør:
obabel -:"SMILES" -O ligand.sdf --gen3D
¶ Metode C - Schrödinger Maestro
- Select Prepared Ligander, se LigPrep
- Right click the structures of interest
- Export
- Chose the format of interest MOL2/SDF
¶ 2) Start systembygging i CHARMM-GUI (Relative Ligand Binder)
Åpne Relative Ligand Binder:
🔗 https://www.charmm-gui.org/?doc=input/afes.rbinding
¶ 🔬 Fremgangsmåte
- Protein: Bruk PDB-ID eller last opp
.pdb(Maestro-preppet er OK).
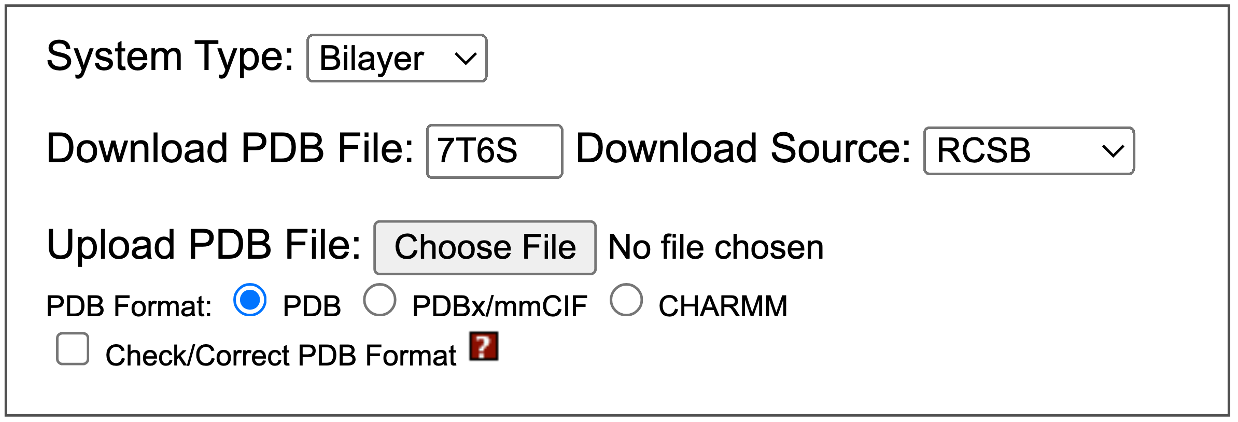
- Kryss av Check/Correct PDB Format.
- Velg kjede og identifiser referanseligand (for eksempel HETA).
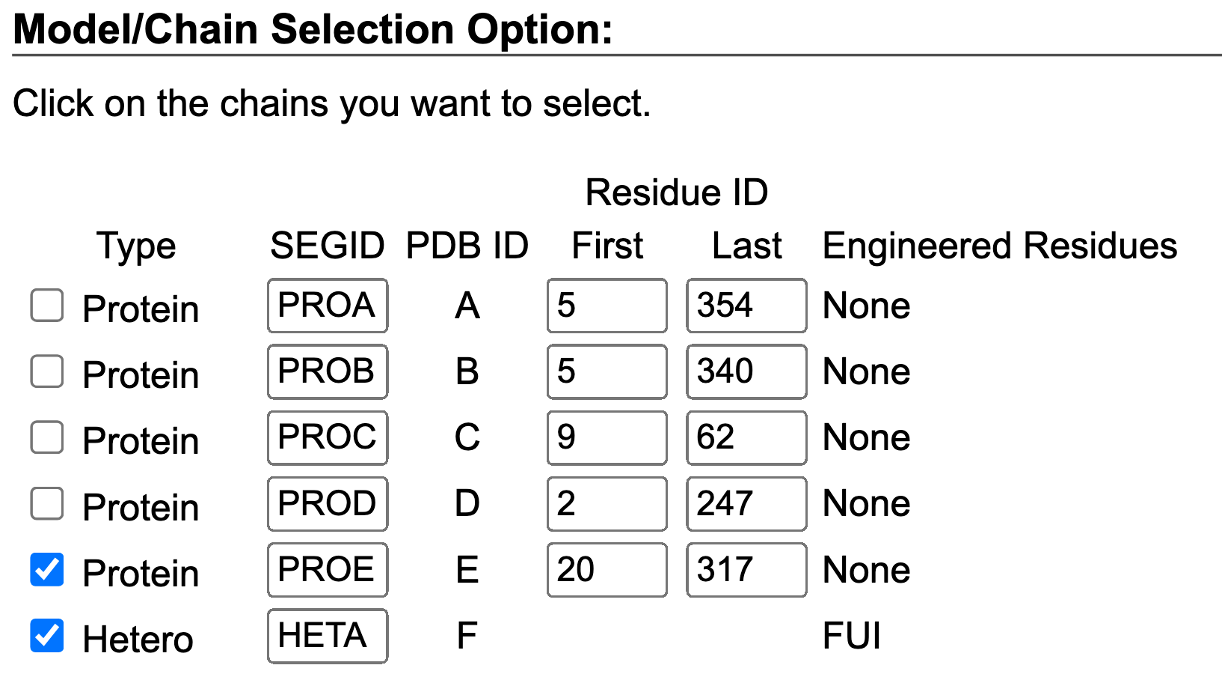
- Angi pH og terminale grupper.
- Last opp
.mol2eller.sdf-fil for referanseliganden.- Hvis bindingsposisjonen til referanseliganden din allerede er bestemt av røntgen/kryo-EM, er det bedre å bruke SDF-filen fra RCSB
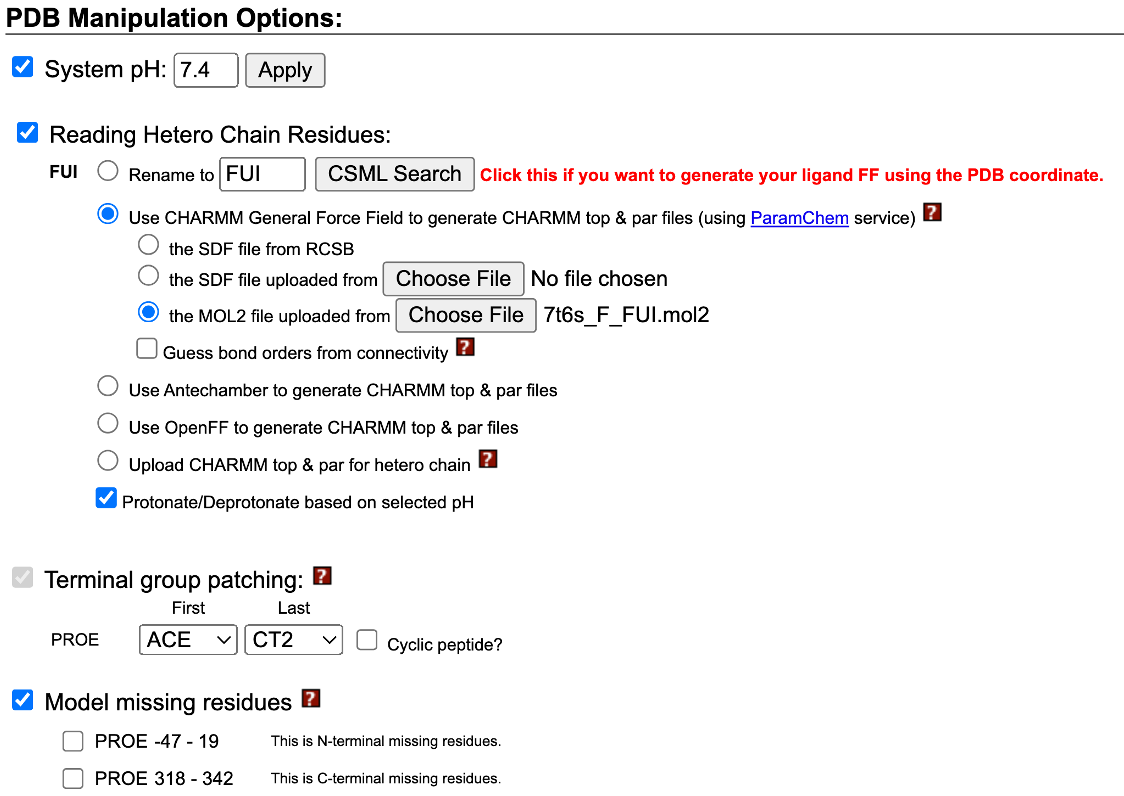
- Hvis bindingsposisjonen til referanseliganden din allerede er bestemt av røntgen/kryo-EM, er det bedre å bruke SDF-filen fra RCSB
- Velg disulfid bonds
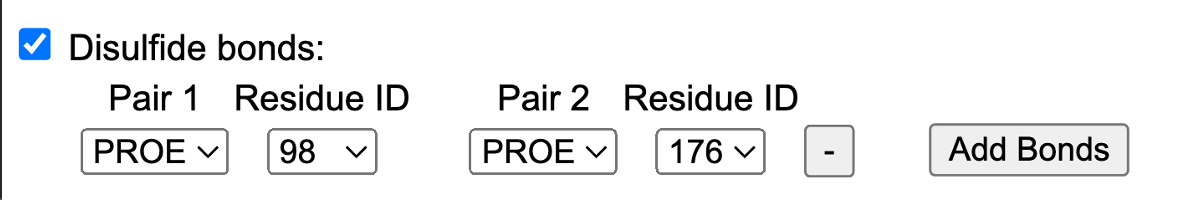
💡 Tips (disulfidbroer):
Sjekk disulfid-broen i UniProt for riktig kobling.
¶ 3) Membranorientering (for 7TM/protein i membran)
Kjør PPM 2.0 (OPM) via CHARMM-GUI for automatisk orientering:
🔗 https://opm.phar.umich.edu/ppm_server2

¶ 4) Membranbygging
-
Velg membran, ofte POPC (1:1) som standard.
- Du kan tilpasse et membran-dobbeltlag for best å etterligne proteinmiljøet ditt, men en POPC-membran med et 1:1-forhold bør være OK.
-
Sett membran dimensjoner med buffer (typisk ~70 Å i X/Y for GPCR).
- 👉 Trenger du hjelp til å finne dimensjoner som passer ditt protein? Se egen tutorial her.
-
Sjekk at orienteringen ser korrekt ut før du fortsetter.
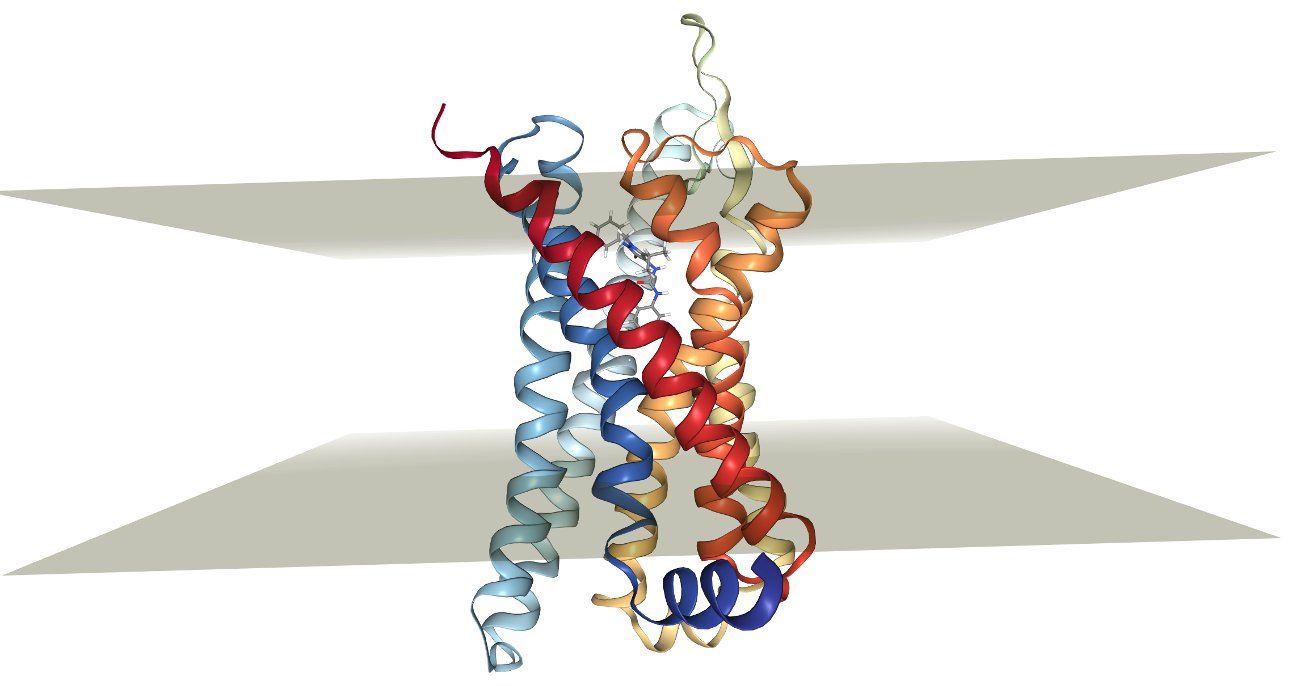
Bilde: Bilayer-konfigurasjon i CHARMM-GUI + orientert 7TM.
¶ 5) Solvatisering og ionisering
- Velg Ion Placing Method og angi ionetype/konsentrasjon.
- Bruk Monte Carlo Method (mer realistisk, men tregere).
- Mer naturlig fordeling av ioner.
- Ulempe: Tregere, siden det innebærer iterativ optimalisering.
- Foretrukket for membran-protein-systemer
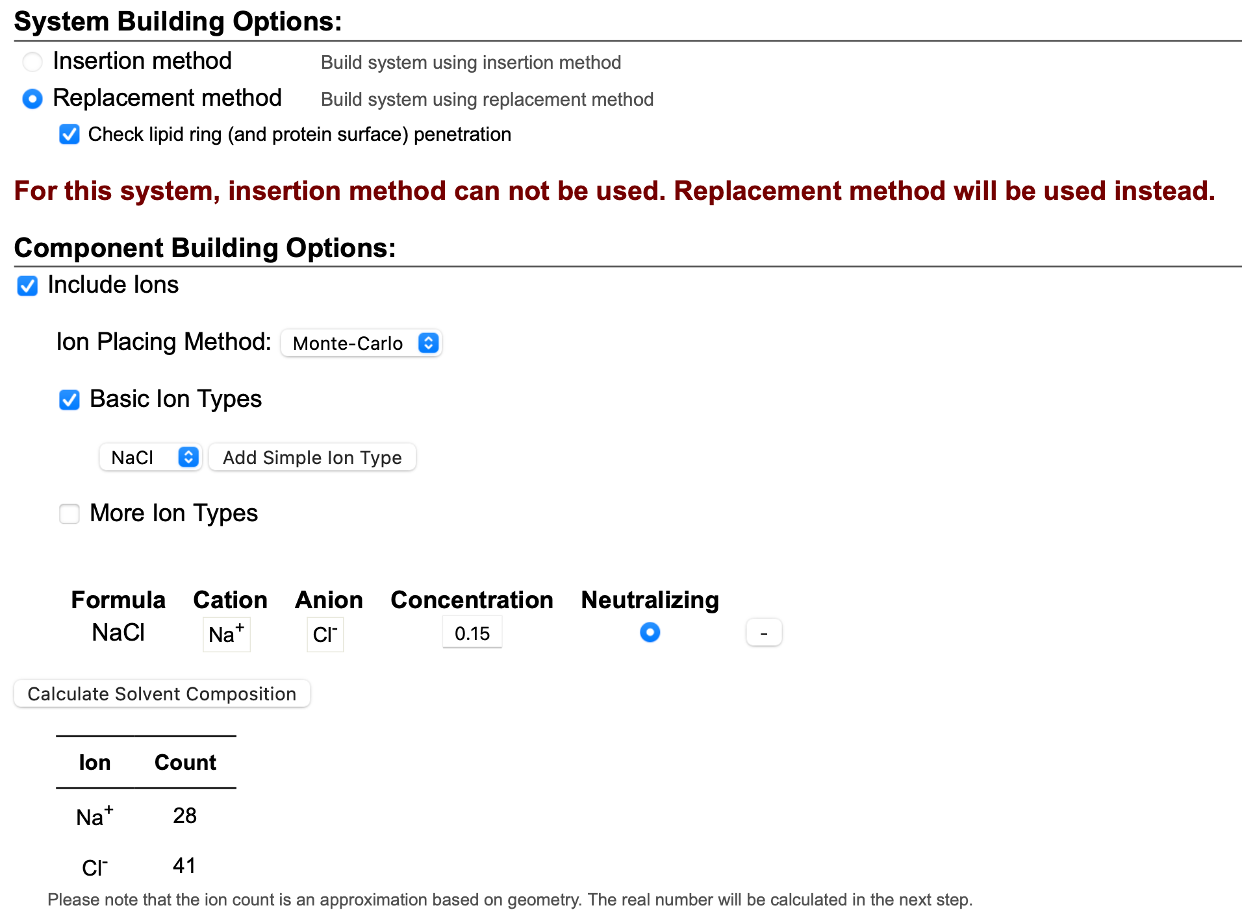
Bilde: Ion Placement-trinnet i CHARMM-GUI.
- Kjør lipid penetration check og kontroller eventuelle kollisjoner.
¶ 6) Legg inn målligand(er) for relativ FEP
- Last opp
.sdf-fil for hver analog. - Velg perturbation path som radial fra referansen (ingen clustering) hvis alle ligander skal sammenlignes direkte mot referanseliganden.
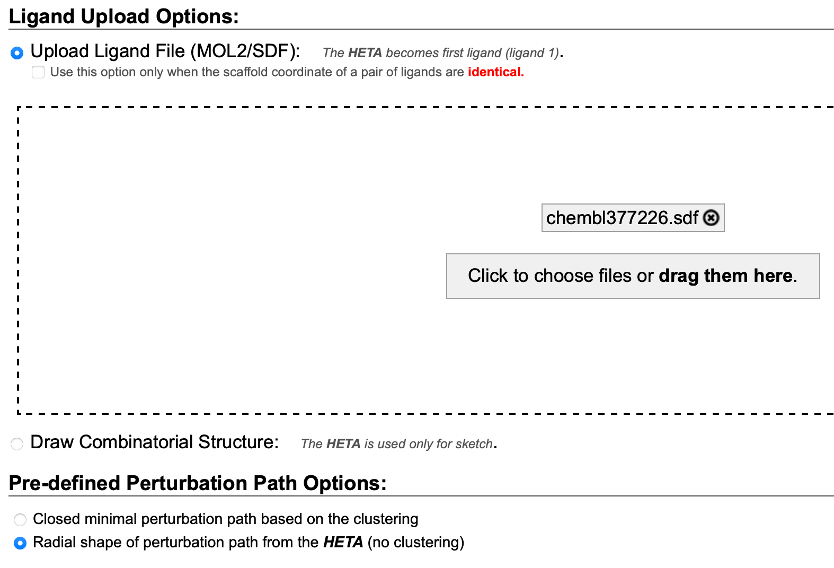
Bilde: “Upload Target Ligands” + valg av radial path.
¶ 7) Oppsett av ligand-par og input-generering
Når du har lastet opp både referanse- og målligandene, går du videre til fanen Pair for å definere
hvilke ligander som skal sammenlignes i den relative FEP-simuleringen.
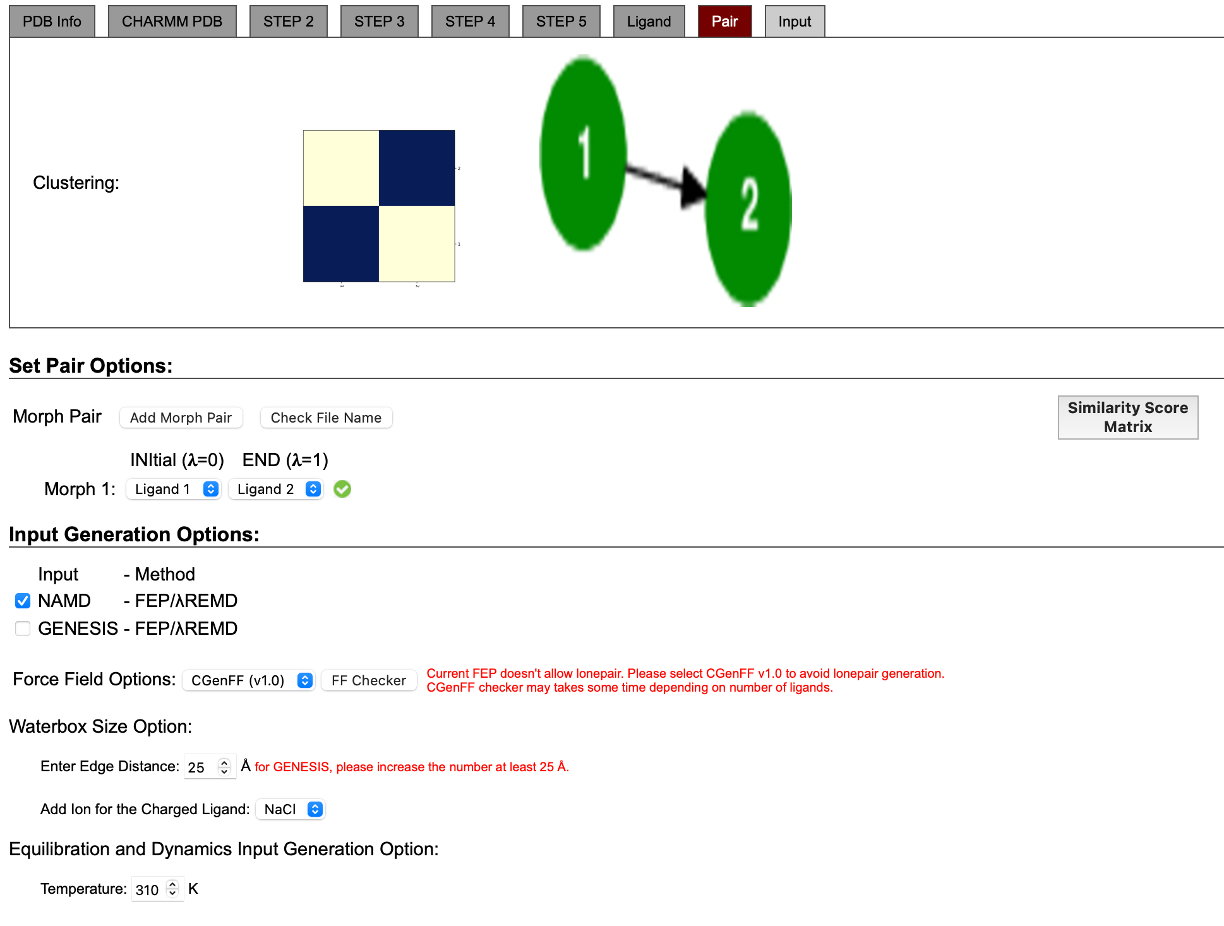
¶ ⚙️ Forklaring av innstillinger
-
Set Pair Options:
- Klikk Add Morph Pair for å opprette et par mellom to ligander.
- Under Initial (λ = 0) velger du referanseliganden (Ligand 1).
- Under End (λ = 1) velger du målliganden (Ligand 2).
- Denne transformasjonen utgjør én FEP-prosess (fra ligand 1 → ligand 2).
💡 I dette prosjektet brukes radial tilnærming, ikke clustering.
Det betyr at hver ligand sammenlignes direkte med én felles referanseligand. -
Input Generation Options:
Her velger du hvilke filer CHARMM-GUI skal generere:- Merk av NAMD → FEP/λ-REMD (standard for denne wikiens eksempler).
- Under Force Field Options, velg CGenFF v1.0 for å unngå lone-pair-feil.
(Rød tekst under feltet minner deg om dette.) - Hvis du velger FF Checker, kan du verifisere atomtyper før generering --> det må vises en grønn hake (✅) ved siden av morph parene.
-
Waterbox Size Option:
- Standard kantlengde er 25 Å.
- I de fleste NAMD-oppsett er 25 Å tilstrekkelig for å unngå interaksjoner over periodiske grenser.
- Legg til ioner hvis systemet er ladet (vanligvis
NaCl).
-
Equilibration and Dynamics Input:
- Angi temperatur (som oftest 310 K, fysiologisk).
- CHARMM-GUI genererer deretter input-filer for energi-minimering, equilibration og λ-REMD.
💡 Tips:
Hvis du har flere ligander, kan du definere flere morph pairs — alle utgående fra
den samme referanseliganden. Dette kalles et radialt FEP-nettverk.
⚠️ NB! Hvis du velger å generere input-filer for flere ligander samtidig,
kan det oppstå mangler eller ufullstendige filer i enkelte systemer.
For å unngå dette anbefales det å generere filer én targetligand av gangen.
¶ 8) Last ned systempakken
Når systemet er ferdig bygget: last ned .tgz-filen.
# Eksempel på opplasting til SAGA
scp charmm.tgz [email protected]:/path/to/project
# Alternativt:
rsync -avzP charmm.tgz [email protected]:/path/to/project
# Pakk ut arkivet:
tar -xvzf charmm.tgz
¶ Neste steg – λ-REMD ved bruk av NAMD
Når systempakken er lastet opp og pakket ut på HPC-serveren,
er du klar til å kjøre simuleringene som ble generert i CHARMM-GUI.
I dette trinnet bruker vi λ-REMD (lambda-Replica Exchange Molecular Dynamics)-metoden,
som gjør det mulig å utveksle replikater mellom ulike λ-verdier for å forbedre sampling og konvergens i FEP-beregningen.
For en detaljert forklaring av hvordan du setter opp, kjører og overvåker simuleringene på HPC, se neste veiledning:
📄 Sist oppdatert: September 2025
👤 Forfatter: Majd Awad
🏫 Farmasøytisk institutt, Universitetet i Oslo
📘 Relatert forskningsgruppe: LIPCHEM